Motivation
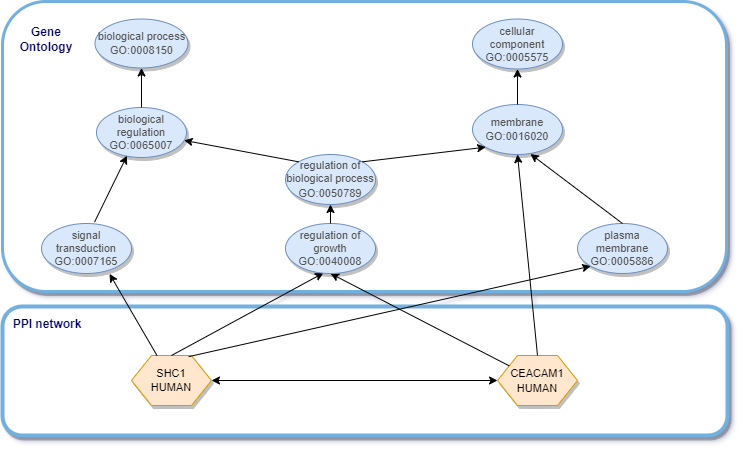
Graphs provide a data structure for knowledge representation and are useful for the description of relationships in all kinds of knowledge domains, including biological systems.
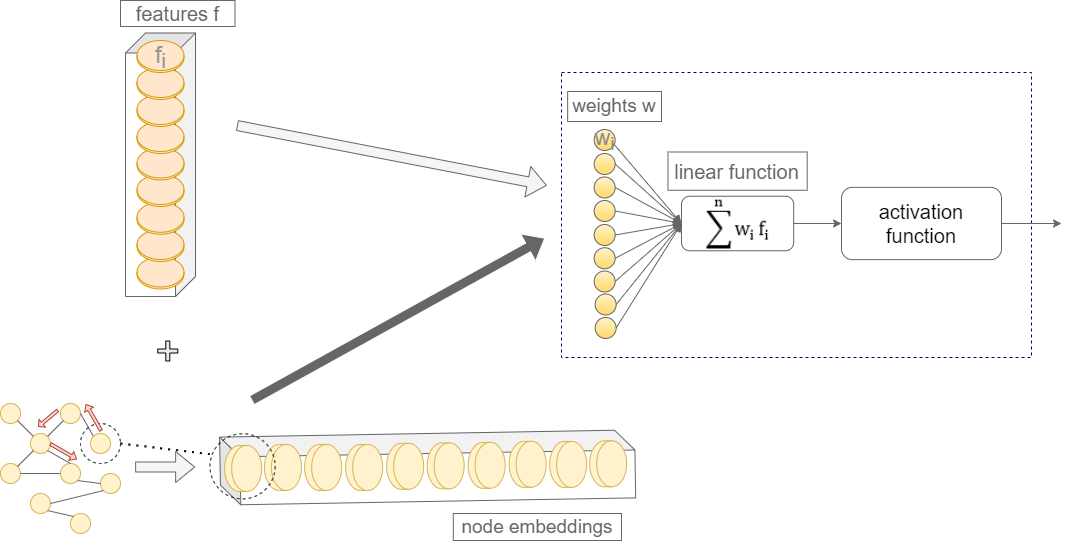
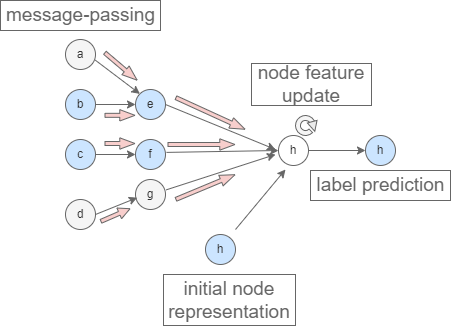
Knowledge graphs (KG) allow for the representation of knowledge through a logical description of its concepts, including of biological entities. By combining biological data with the relevant biological knowledge available, we’ll be bringing knowledge graphs into data mining tasks, and creating an opportunity for ML models to learn from the enriched graph data.